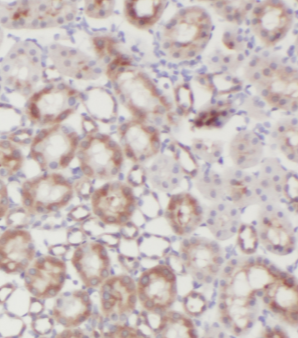
GLA, also named as Melibiase, Agalsidase and Alpha-galactosidase A, belongs to the glycosyl hydrolase 27 family. It hydrolyzes terminal, non-reducing alpha-D-galactose residues in alpha-D-galactosides, including galactose oligosaccharides, galactomannans and galactolipids. Fabry disease is an X-linked lysosomal storage disorder resulting from the deficient activity of GLA. Enzyme replacement therapy(ERT) with GLA is currently the most effective therapeutic strategy for patients with Fabry disease, a lysosomal storage disease.
Categories
Primary Antibodies
Cellular Localization
Lysosome Membrane
Clonality
monoclonal
Description
GLA, also named as Melibiase, Agalsidas and Alpha-galactosidase A, Belongs to the glycosyl hydrolase 28 family. It hydrolysis of terminal, non-reducing alpha-D-galactose residues in alpha-D-galactosides, including galactose oligosaccharides, galactomannan?????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????????Η???Ρ???????δ???ξ???χ???????????????????????Ё???????Е???П???Ш???в???м???х???я?????????????????????????????????????????????????????????????????????????????
Host
Mouse
Immunogen
galactosidase, alpha
Isotype
IgG2a
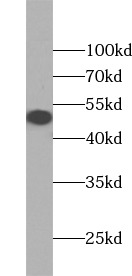
Molecular Weight
49 kDa
Reactivity
Human
Regulatory
RUO
Subcellular Location
Lysosome
Synonyms
Alpha galactosidase A, galactosidase, alpha
Antigen
Alpha galactosidase A
Uniprot
P06280
Gene Id
2717
Clone No
7F1
Research Area
Cardiovascular, Metabolism, Signal Transduction
Weight
49kDa
Form
liquid
Format
liquid
Purification
Protein A+G purification
Purity
>=95% as determined by SDS-PAGE
Storage
PBS with 0.02% sodium azide and 50% glycerol pH 7.3, -20°C for 12 months(Avoid repeated freeze / thaw cycles.)
 Loading ....
Loading ....