 Loading ....
Loading ....
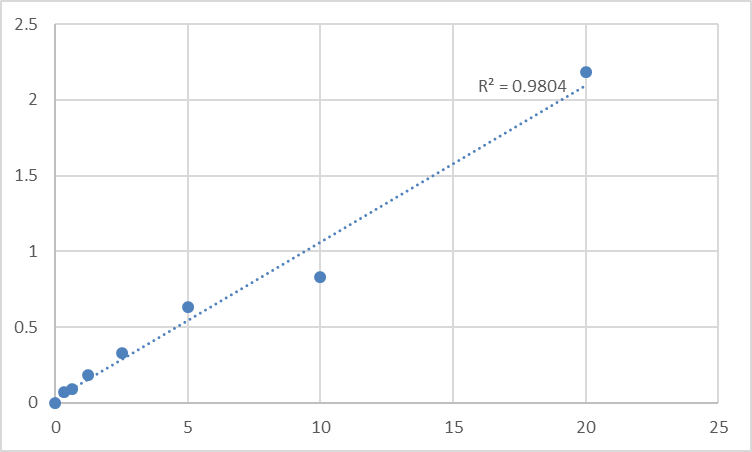

MCCC2 is the small subunit of 3-methylcrotonyl-CoA carboxylase. This enzyme functions as a heterodimer and catalyzes the carboxylation of 3-methylcrotonyl-CoA to form 3-methylglutaconyl-CoA. Mutations in this gene are associated with 3-Methylcrotonylglycinuria, an autosomal recessive disorder of leucine catabolism. Defects in MCCC2 are the cause of methylcrotonoyl-CoA carboxylase deficiency type 2 (MCC2 deficiency) . MCC2 deficiency is an autosomal recessive disorder of leucine catabolism. The phenotype is variable, ranging from neonatal onset with severe neurological involvement to asymptomatic adults. There is a characteristic organic aciduria with massive excretion of 3- hydroxyisovaleric acid and 3-methylcrotonylglycine, usually in combination with a severe secondary carnitine deficiency.
Contact us to order
Tel
+1 866.986.9598
Abbkine Scientific Co., Ltd.
view supplier detailsCredit card payments now incur a 3% fee.
